Introduction
Ginkgo’s mission is to make biology easier to engineer, so the ability to read the DNA of the organisms that we engineer at scale is critical to the success of our mission. Sequencing DNA at scale, commonly known as Next Generation Sequencing (NGS), enables Ginkgo Bioworks to sequence thousands of samples per day, and is central to Concentric, Ginkgo Bioworks’ COVID-19 diagnostics platform. An enormous amount of data comes out of our sequencing instruments each day, and we have built data processing pipelines to turn these terabytes of raw sequencer data into actionable insights for our scientists. Here we offer a history and discussion of some of the design decisions we made in the development of this pipeline.
Some Background
To understand the use of NGS at Ginkgo, it helps to understand how it is traditionally used. When engineering organisms, a scientist will often try to modify an organism’s DNA to change some functionality. For instance, they may want to insert a gene that makes the organism fluoresce. However, this process is often non-perfect and the inserted DNA can have errors in it, be inserted into an unintended region of the genome, be inserted backwards, and many more fun possibilities. NGS helps answer the question of “what did I actually do” by providing the sequence so the scientist can compare their intended engineering with the actual engineering.
To facilitate this, Ginkgo employs a variant of NGS known as short read sequencing, where larger molecules of DNA are fragmented into short segments. These short segments are then mapped against the intended genome so errors and discrepancies can be discovered. Here’s how some real data looks, using IGV to visualize the alignments. The top bar is the genomic sequence, and each grey bar represents a genomic fragment read off a sequencer. The mismatches between the genome and the segment is shown when a letter appears in the segment.
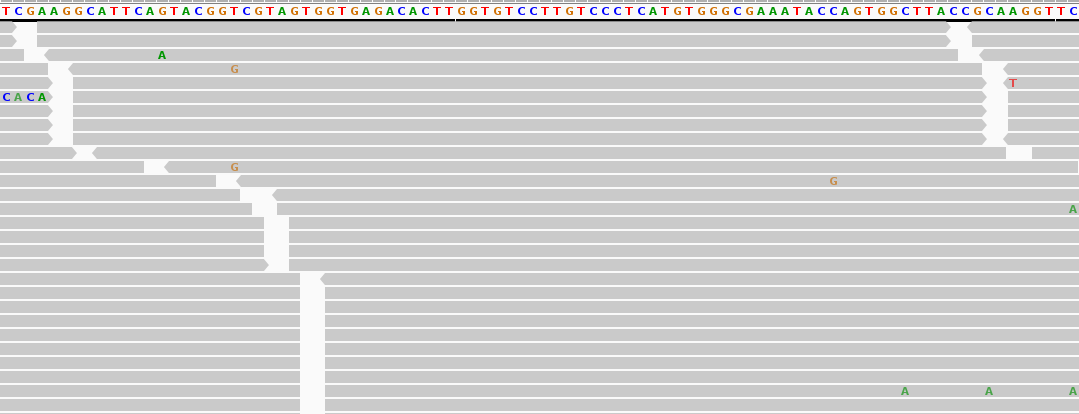
This data is actually from our COVID19 Whole Genome Sequencing service, where we track the mutations and spread of different SARS-CoV-2 strains to help develop diagnostics and contain its spread.
Multiplexing NGS
Although sequencers can produce billions of data points, a given sample usually does not need that much data. Thus, we often want to divide the sequences among a number of samples. This technique is known as multiplexing. In NGS, this is accomplished by attaching a short, known genetic sequence to each end of a DNA fragment known as a barcode. In this diagram, each unique barcode is represented as a color.
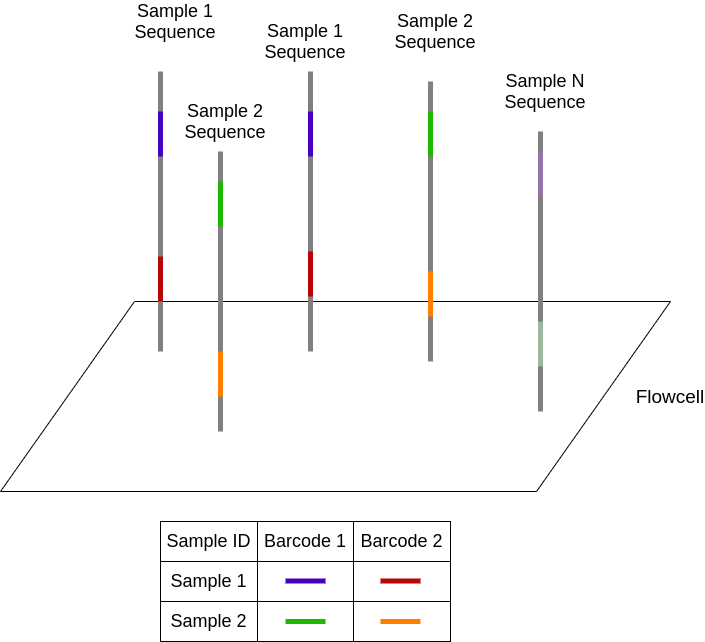
During sequencing, the barcodes are read out and can be mapped back to the sample ID prior to mixing. This process of assigning the generated reads back to each individual sample is known as demultiplexing. In most cases, a few dozen samples are sequenced in parallel. At Ginkgo, we regularly run thousands of samples in parallel due to our amazing automation capabilities that can perform the immense amount of liquid transfers required to multiplex to this extent. As you can imagine, this generates a ton of data – let’s take a look at how we process it.
Version 1.0 with Celery
The initial NGS Pipeline was based on Celery Canvas, a Python task workflow framework. This is a dynamic system where a task would provision the next stages based on the current task (e.g. if a reference genome was known, a workflow for reference based alignment would be performed). It was insanely fast with respect to task orchestration overhead. Thousands of tasks would execute across the cluster in seconds without needing any database queries to manage the state of a workflow or tasks. Here is a very simple diagram of this infrastructure, with a simple workflow:
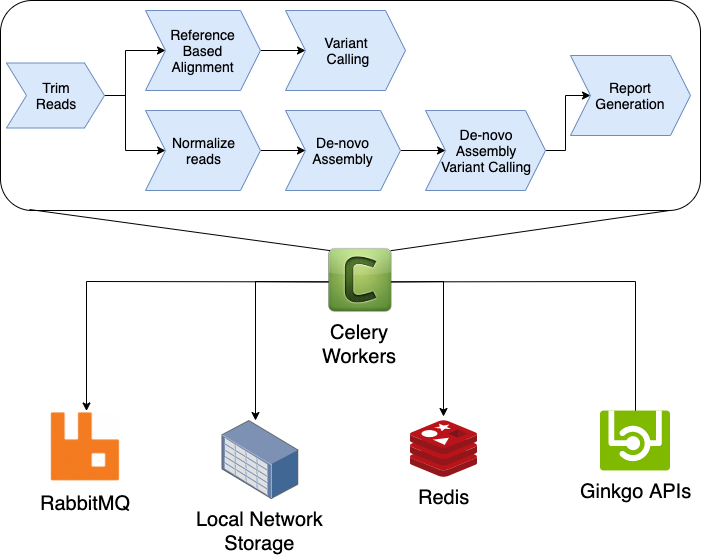
However, as Ginkgo’s sequencing capabilities and scale grew, we encountered shortcomings with this architecture.
Shortcomings
Error Handling
The NGS Pipeline depended on a number of internal services with varying levels of robustness and deployment strategies. Thus, we had to handle cases where a server was down because it was being deployed, or it simply crashed under the storm of NGS related API calls.
Managing this is non-trivial with Celery, as it requires the user to explicitly handle error cases for retries ahead of time, without any manual curation or intervention when the event does happen. One option is to implement very liberal exception handling, where Celery essentially retries for a given period of time until quitting, but sometimes this masks a very real error, and the developer has to go and manually kill the task, or try to steal it out of the broker to stop the application. We took a conservative approach, defining the error modes as we encountered them, and then restarting the NGS Pipeline to re-analyze results.
This brings us to our next problem, reusing previous work.
Results Reuse
Reusing previous results is non-trivial, and is not something Celery natively supports (for good reasons, it’s a hard problem that is application dependent). We had a need to reuse results because some tasks could take on the order of hours to complete, so we wrote a custom Celery task that attempted to cleverly hash the inputs and cache results based on those hashes. The reality, though, was that the types of inputs into the system and the sources of those inputs varied widely, so the caching introduced a great deal of complexity. Humans are a lot better at knowing when a cache should be used or not. Caching is hard.
Introspection
Introspection was also a problem. Our users would sometimes ask “Where’s my data?” or “What is the status of my request?” There are ways to introspect Celery task status from the command line, but these are obviously not a self-service interfaces for our users. There are UIs for Celery task monitoring (such as Flower and a home-grown one), but they are often missing features or added complexity and points of failure. We did not want NGS Pipeline development to grow in scope to the point where we were building and maintaining task orchestration and monitoring UIs.
Resource Usage
As the NGS Pipeline grew to new domains and analyses, such as de-novo assembly, we began hitting resource constraints. Celery does not natively offer a way to refuse or allocate tasks based on the host resource availability and usage. Some work was done on this as well, extending our custom Celery task to reject a task if the host could not accommodate the workload. This worked, but bouncing tasks around workers until one was found that could perform the work is inefficient.
Re-architecting the Pipeline
Celery allowed us to stand up a high-throughput pipeline in an incredibly short time, and taught us valuable lessons about what features were essential to a workflow engine at Ginkgo. However, it was clear we were at the junction where addressing all our needs would be inventing a new workflow orchestration service. Thus, it was clear that we had to re-architect it and choose different technologies in order to meet our needs. In my next blog post, I will tell you which technologies we chose and how they enabled us to build a more robust NGS Pipeline.
(Feature photo by Christophe Dion on Unsplash)
